Highly optimized Burrow-Wheeler Aligner specifically for Illumina ~150 bp short-read alignment.
Hewill Aligner is a highly optimized Burrow-Wheeler Aligner (implement using modern C++20) specifically for paired-end short read alignment with read length ~150 bp.
The elapsed time of mapping sequencing data with 50× coverage (2×180GB) is less than 2 hours under 80 cores and ~18 GB memory usage and has comparable performance to bwa-mem2.
sudo apt install libtbb-dev)
$ g++-10 main.cpp -o hewill -pthread -ltbb -std=c++20 -O3 biomodern/ssw.cpp -Wno-ignored-attributes
Command:./hewill index <hs37d5.fa>./hewill align <hs37d5.fa> <in1.fq> <in2.fq> <sam_prefix> <sample_name> <read_group_id> [insert_mean] [insert_var] [thread_num]Required Arguments:<hs37d5.fa> Reference sequence hs37d5.fa file path<in1.fq> Read1 FASTQ path<in2.fq> Read2 FASTQ path<sam_prefix> SAM file output prefix<sample_name> SAM file SM tag value<read_group_id> SAM file RGID tag valueOptional Arguments:[insert_mean] Mean insert size of the paired-end data, default value is 550[insert_var] Variance insert size of the paired-end data, default value is 150[thread_num] Number of threads, default value is return value of std::thread::hardware_concurrency()
$ ./hewill index /mnt/fa/hs37d5.fa
// <hs37d5.fa> <in1.fq> <in2.fq> <sam_prefix> <sample_name> <read_group_id> [insert_mean] [insert_var] [thread_num]$ ./hewill align /mnt/fa/hs37d5.fa /mnt/fq/HG001.1.fq /mnt/fq/HG001.2.fq /mnt/sam/HG001 HG001 1 550 150 80
PrecisionFDA Truth Challenge benchmark versus bwa-mem2: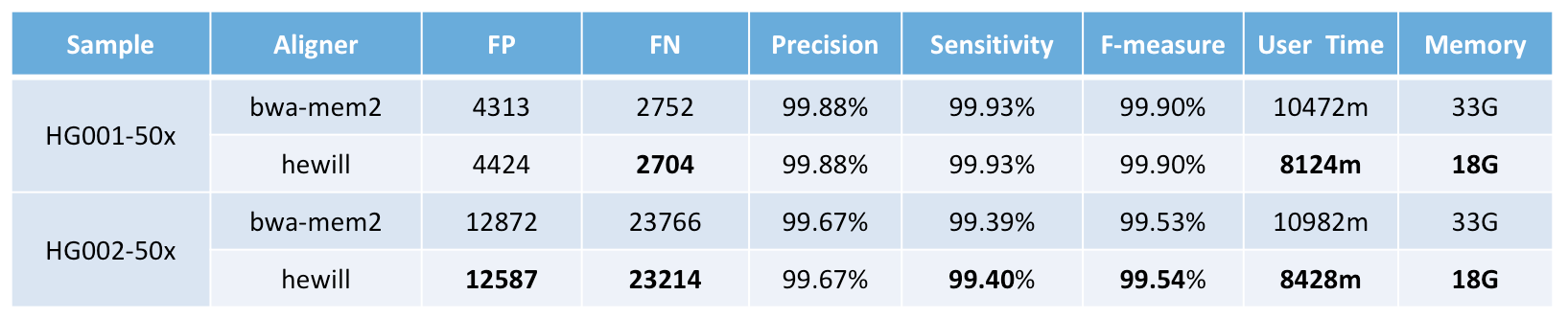
This aligner is highly optimized on the following sequencing characteristic (other datasets are not recommended):
If you know the mean and variance of the insert size of the sequencing data, we highly recommend you pass it into the aligner.